Aansprakelijkheid na tuchrechtprocedure: stuiting verjaring
Hof Arnhem 11 september 2012, LJN BX6567 (informed consent ontwikkelingsachterstand)

Aansprakelijkheid arts (psychiater) voor voorschrijven classificatie-C medicijnen tijdens eerste weken zwangerschap? Informed consent? Aansprakelijkstelling na tuchtrechtelijke procedure bij het Regionaal Tuchtcollege te Amsterdam (RTC). Verjaring. Stuiting door klaagschrift. Causaal verband medicijngebruik en ontwikkelingsachterstand (psychomotore retardatie) van de op 15 juli 1998 geboren zoon. Medicamenteuze beleid van [X] op zich niet onverdedigbaar, maar er is wel onzorgvuldig gehandeld.
De grieven 5 en 6 slagen. De niet op 20 augustus 1998 maar eerst in 2001 aangevangen verjaring is in ieder geval gestuit door de stellingen in het klaagschrift en de repliek van 30 augustus 2005 in de tuchtzaak, waarna de aansprakelijkstelling van 26 oktober 2007 en de dagvaarding van 21 juli 2009 zijn gevolgd. Het verjaringsverweer wordt verworpen.
De devolutieve werking van het hoger beroep brengt mee dat het hof alsnog de genoemde vorderingen inhoudelijk dient te beoordelen.
Uit de uitspraak van het RTC blijkt dat het college, gezien de ernstige problemen waarmee [moeder] in 1998 kampte, het medicamenteuze beleid van [X] op zich niet onverdedigbaar oordeelt. Wel heeft [X] onzorgvuldig gehandeld door de in het Repertorium en het Kompas genoemde risico's niet met patiënte te bespreken voordat hij de medicijnen voorschreef en door haar uitsluitend gerust te (willen) stellen onvolledige informatie heeft verschaft. Hij had samen met de patiënte en klager het nut van de medicatie voor de patiënte moeten afwegen tegen de risico's van het kind. Een dergelijk gesprek had, zeker nu beide middelen onder de categorie C geneesmiddelen vallen, aanleiding moeten zijn voor extra voorzichtigheid, aldus het RTC.
Het hof heeft behoefte aan voorlichting van één of meer deskundigen (het hof denkt vooralsnog aan een psychiater en een farmacoloog) teneinde antwoord te krijgen op de vraag of [X] in 1998 als redelijk handelend en redelijk bekwaam psychiater heeft gehandeld.
3.2 Op 15 januari 1998 heeft [X] [moeder] tijdens zijn spreekuur gezien. Op grond van zijn bevindingen en diagnose (gejaagd depressief syndroom), die zijn vastgelegd in de patiëntenkaart, heeft hij haar de geneesmiddelen rivotril en anafranil voorgeschreven. Bij brief van 18 januari 1998 heeft [X] [gynaecoloog] en de huisarts van [moeder] hierover geïnformeerd.
4.1. Appellanten houden [X] aansprakelijk voor de geestelijke en lichamelijke handicaps van [zoon]. Zij stellen dat [X] door [vader]- in dat stadium van haar zwangerschap deze medicijnen voor te schrijven jegens [moeder] en [zoon] in zijn zorg- en informatieplicht is tekortgeschoten in de nakoming van de geneeskundige behandelingsovereenkomst en jegens [vader] onrechtmatig heeft gehandeld.
4.7 Het hof deelt de overweging van de rechtbank (vonnis onder 4.6) dat de aard en strekking van een procedure voor de tuchtrechter voor beroepsbeoefenaren (in dit geval: de medische tuchtrechter) van andere aard is dan een aansprakelijkheidsprocedure voor de burgerlijke rechter en dat een procedure voor die tuchtrechter niet tot doel heeft de civielrechtelijke aansprakelijkheid van de beroepsbeoefenaar vast te stellen. Een en ander sluit echter niet uit dat in de door een klager voor de tuchtrechter ingediende stukken en gevoerde betogen “een schriftelijke mededeling waarin de schuldeiser zich ondubbelzinnig zijn recht op nakoming van een verbintenis voorbehoudt” in de zin van art 3:317 lid 1 BW besloten ligt. Naar vaste rechtspraak van de Hoge Raad moeten de aangehaalde woorden begrepen worden in het licht van de strekking van deze stuitingshandeling, die erop neerkomt dat er sprake moet zijn van een voldoende waarschuwing aan de schuldenaar dat hij rekening ermee moet houden dat hij ook na het verstrijken van de verjaringstermijn de beschikking houdt over zijn gegevens en bewijsmateriaal, opdat hij zich tegen een dan mogelijkerwijs alsnog ingestelde rechtsvordering behoorlijk kan verweren. In de onderhavige zaak zal daarom, gelet ook op hetgeen appellanten hebben aangevoerd, de vraag moeten worden beantwoord of de door [vader] bij het Regionaal Tuchtcollege ingediende stukken een voldoende duidelijke waarschuwing aan [X] behelsden, dat hij rekening ermee moest houden dat hij ook na het verstrijken van de verjaringstermijn de beschikking houdt over zijn gegevens en bewijsmateriaal, opdat hij zich tegen een dan mogelijkerwijs alsnog in te stellen rechtsvordering behoorlijk kan verweren.
4.8 Die vraag beantwoordt het hof, anders dan de rechtbank, bevestigend.
4.9 De devolutieve werking van het hoger beroep brengt mee dat het hof alsnog de onder 4.2 genoemde vorderingen inhoudelijk dient te beoordelen. Appellanten houden [X] aansprakelijk voor de (gezondheids)schade van [zoon], omdat hij tekortgeschoten is in zijn zorg- en informatieverplichting, hetgeen volgens hen als een toerekenbare tekortkoming en/of als onrechtmatig handelen kan worden gekwalificeerd. Uit de stellingen van appellanten in eerste aanleg en in hoger beroep leidt het hof af dat zij niet alleen aanvoeren dat [X] zijn informatieverplichting betreffende het al dan niet voorschrijven van de medicatie heeft geschonden, maar ook dat [X] zijn zorgverplichting heeft geschonden door twee geneesmiddelen voor te schrijven met classificatie C, geneesmiddelen waarvan bekend is of moet worden vermoed dat zij door hun farmacologische effecten stoornissen bij het ongeboren kind kunnen veroorzaken.
Uit de uitspraak van het RTC blijkt dat het college, gezien de ernstige problemen waarmee [moeder] in 1998 kampte, het medicamenteuze beleid van [X] op zich niet onverdedigbaar oordeelt. Wel heeft [X] onzorgvuldig gehandeld door de in het Repertorium en het Kompas genoemde risico's niet met patiënte te bespreken voordat hij de medicijnen voorschreef en door haar uitsluitend gerust te (willen) stellen onvolledige informatie heeft verschaft. Hij had samen met de patiënte en klager het nut van de medicatie voor de patiënte moeten afwegen tegen de risico's van het kind. Een dergelijk gesprek had, zeker nu beide middelen onder de categorie C geneesmiddelen vallen, aanleiding moeten zijn voor extra voorzichtigheid, aldus het RTC.
4.10 Het hof heeft behoefte aan voorlichting van één of meer deskundigen (het hof denkt vooralsnog aan een psychiater en een farmacoloog) teneinde antwoord te krijgen op de vraag of [X] in 1998 als redelijk handelend en redelijk bekwaam psychiater heeft gehandeld door anafranil en rivotril aan [moeder] voor te schrijven tijdens haar zwangerschap en over de vraag of er (volledig) causaal verband bestaat tussen de gezondheidsschade van [zoon] en het gebruik van voornoemde middelen door [moeder] tijdens haar zwangerschap.
Voor dit doeldier is geen Maximum Residu Limiet bepaald
Rechtbank Roermond 13 september 2012, LJN BX7525 (Geen toepassing cascaderegeling)
 Diergeneesmiddelenwet art. 2 + Diergeneesmiddelenbesluit art. 23, 24. Dierenwelzijn: diergeneesmiddelen van goede kwaliteit tegenover voedselproducerende dieren voor het belang van de volksgezondheid. Diergeneeskundige noodzaak, maar geen noodtoestand. Geen toepassing cascaderegeling geen Maximum Residu Limiet bepaald voor kalkoen.
Diergeneesmiddelenwet art. 2 + Diergeneesmiddelenbesluit art. 23, 24. Dierenwelzijn: diergeneesmiddelen van goede kwaliteit tegenover voedselproducerende dieren voor het belang van de volksgezondheid. Diergeneeskundige noodzaak, maar geen noodtoestand. Geen toepassing cascaderegeling geen Maximum Residu Limiet bepaald voor kalkoen.
Verdachte is tenlastegelegd dat hij op 26 januari 2011 te Panningen, in de gemeente Peel en Maas, althans in Nederland, al dan niet opzettelijk, drie- althans eenmaal, (telkens) een diergeneesmiddel dat niet was geregistreerd (voor pluimvee) heeft afgeleverd en/of bij dieren (pluimvee) heeft toegepast (...)
Officier van Justitie: Uit de toepasselijke regelgeving met name artikel 23 Diergeneesmiddelenbesluit en de in dat artikel genoemde verordening (EG) nr. 2377/90 vloeit voort dat het middel ceftiofur (de werkzame stof in Excenel) niet is toegestaan voor het doeldier kalkoen, nu voor dit doeldier geen Maximum Residu Limiet (MRL) is bepaald.
Verweer: Zakelijk weergegeven is aangevoerd dat er sprake was van een diergeneeskundige noodzaak zoals bedoeld in artikel 22 van het Diergeneesmiddelenbesluit en dat de zogenaamde cascaderegeling van de artikelen 22 tot en met 24 van het Diergeneesmiddelenbesluit correct is toegepast. Immers in dit kader is toepassing van een toegelaten geneesmiddel voor een andere voedselproducerende diersoort toegestaan.
Rechter: Met betrekking tot de uitleg van de cascaderegeling overweegt de economische politierechter als volgt. De strekking van de onderhavige wet- en regelgeving is tweeledig. Enerzijds dienen in het belang van het dierenwelzijn alleen diergeneesmiddelen te worden toegelaten die van goede kwaliteit zijn. Anderzijds is er in het geval van voedselproducerende dieren ook nog het belang van de volksgezondheid. Voorkomen dient immers te worden dat via de consumptie van dierlijke voedingsstoffen en de daarin achtergebleven (medicinale) stoffen de gezondheid van de mens op korte of op de langere termijn nadelig wordt beïnvloed. In het geval van voedselproducerende dieren dient een redelijke wetsuitleg dan ook altijd te geschieden tegen de achtergrond van het belang van de bescherming van de volksgezondheid, in casu met name het gevaar van de toenemende antibioticaresistentie.
Uit de tekst van de artikelen 22 en 23 van het Diergeneesmiddelenbesluit volgt voorts dat het bepaalde in artikel 22 niet los gezien kan worden van het bepaalde in artikel 23. Met andere woorden: men komt pas toe aan toepassing van artikel 22 indien aan de voorwaarden van artikel 23 is voldaan. Dit brengt met zich mee dat de economische politierechter met de officier van justitie van oordeel is dat het gebruik van de stof ceftiofur enkel is toegestaan in het geval er voor de desbetreffende diersoort een MRL is bepaald.
Nu de toepassing van ceftiofur, ook in het geval van een diergeneeskundige noodzaak, niet is toegestaan, is de vraag of er in dezen al dan niet sprake was van een diergeneeskundige noodzaak en verdachte derhalve terecht de cascaderegeling heeft menen toe te kunnen passen niet relevant.
Overwegingen van rechtbank
Verdachte heeft uitvoerig aangevoerd waaruit voor hem de diergeneeskundige noodzaak bestond, maar heeft onvoldoende gesteld om van een noodtoestand zoals hierboven bedoeld te kunnen spreken. In dit kader stelt de economische politierechter voorop dat enkel een grotere uitval dan normaal als zodanig geen noodtoestand vormt.
Uit de toelichting van verdachte op zijn beslissing een niet geregistreerd diergeneesmiddel toe te passen concludeert de rechtbank dat deze mede zijn ingegeven zijn door efficiency redenen. Naast een beter gezondheidsresultaat voor de dieren zijn er een aantal praktische voordelen verbonden aan Excenel. Namelijk toediening van Excenel is te combineren met gelijktijdige toediening van het TRT vaccin. Injectie met Excenel kan bij eendagskuiken machinaal geschieden, gelijktijdig met het snavelkappen en de toediening van de TRT vaccin. Het beroep op een noodtoestand wordt dan ook verworpen.
De beslissing is gegrond op de artikelen:
Wetboek van Strafrecht art. 9a, 91
Wet op de economische delicten art. 1, 2, 6,
Diergeneesmiddelenwet art. 2
Diergeneesmiddelenbesluit art. 22, 23
De verlening van het mono-ABC vs combinatie-ABC
Vzr. Rechtbank 's-Gravenhage 14 september 2012, zaaknr. 425814 / KG ZA 12-905 (Sanofi S.A. tegen Pharmachemie B.V. en Teva Pharma) en 426135 / KG ZA 12-928 (Sanofi S.A. tegen Teva Nederland B.V.)
 In't kort: Aanvullend Beschermingscertificaat. Het combinatie-ABC 990006 ziet op irbesartan, desgewenst in de vorm van een zout en/of een hydraat, en hydrochloorthiazide (ook wel aangeduid als HCTZ) en is van kracht tot en met 14 oktober 2013. Het basisoctrooi EP0454511: N-gesubstitueerde heterocyclische derivaten, de bereiding hiervan, farmaceutische preparaten die deze bevatten.
In't kort: Aanvullend Beschermingscertificaat. Het combinatie-ABC 990006 ziet op irbesartan, desgewenst in de vorm van een zout en/of een hydraat, en hydrochloorthiazide (ook wel aangeduid als HCTZ) en is van kracht tot en met 14 oktober 2013. Het basisoctrooi EP0454511: N-gesubstitueerde heterocyclische derivaten, de bereiding hiervan, farmaceutische preparaten die deze bevatten.
Teva stelt dat het combinatie-ABC nietig is en voert daartoe navolgende gronden aan:
i) het combinatie-ABC is verleend in strijd met artikel 3(a) van de (destijds geldende) Verordening (EG) nr. 1768/92 van de Raad van de Europese Gemeenschappen van 18 juni 1992 betreffende de invoering van een aanvullend beschermingscertificaat voor geneesmiddelen (hierna: ABC-Verordening) omdat (a) het product niet wordt beschermd door EP 511 en (b) het ABC is afgegeven in strijd met het doel en de strekking van de ABCVerordening;
ii) het combinatie-ABC zou zijn verleend in strijd met artikel 3(c) van de ABC-Verordening omdat voor irbesartan eveneens met EP 511 als basisoctrooi reeds een ABC was afgegeven;
iii) EP 511 ontbeert inventiviteit.
Aanvullend criterium: In de ABC-Verordening noch in de uitspraken van het Europese Hof van Justitie is evenwel een aanwijzing te vinden voor de door Teva bepleite regel dat los van toetsing aan de vereisten van artikel 3 ABC-Verordening, als aanvullend criterium heeft te gelden dat een ABC in overeenstemming dient te zijn met doel en strekking van de ABC-Verordening.
Een ABC per product per basisoctrooi:
4.19 De voorzieningenrechter merkt voorts op dat algemeen wordt aangenomen dat uit het in 1995 gewezen Biogen-arrest de regel volgt dat (slechts) één ABC kan worden verkregen per product per basisoctrooi. Gelet op het feit dat het in die zaak ging om één geneesmiddel dat door meerdere basisoctrooien wordt beschermd (waarvoor verschillende octrooihouders afzonderlijke ABC’s hadden aangevraagd) en niet om een basisoctrooi dat meerdere producten in de zin van de ABC-Verordening beschermt, kan uit de door het HvJ EU gebruikte formulering dat “voor ieder basisoctrooi niet meer dan één certificaat [mag] worden afgegeven” naar voorlopig oordeel niet worden geconcludeerd dat deze regel zonde meer van toepassing is op een basisoctrooi dat meerdere producten onder bescherming stelt (zoals EP 511).
Er wordt Pharmachemie en Teva Nederland bevolen geen inbreuk te maken op de combinatie-ABC van Sanofi. Tevens wordt ook opgave door registeraccountant en een brief aan alle afnemers toegewezen, onder last van dwangsommen.
In citaten:
ad i) artikel 3 (a) ABC-Verordening
4.11. Het standpunt van Teva dat de afgifte van het combinatie-ABC in strijd zou zijn met doel en strekking van de ABC-Verordening, omdat de tweede marktvergunning (voor de combinatie) ten opzichte van de marktvergunning voor irbesartan – die volgens Teva ook al (mede) zag op de verhandeling van de combinatie van irbesartan met HCTZ – in wezen slechts de toedieningsvorm in één tablet toevoegt, kan niet als juist worden aanvaard. Dat is alleen al zo omdat – naar Teva erkent – het combinatiepreparaat irbesartan / HCTZ volgens de definitie van “product” in de zin van de ABC-Verordening heeft te gelden als een ander product (en niet slechts als andere toedieningsvorm) dan irbesartan, waarvoor ook een aparte marktvergunning is vereist. De verwijzing door Teva naar paragraaf 11 van het Explanatory Memorandum – waarin is opgemerkt dat een nieuwe toedieningsvorm van hetzelfde product geen aparte ABC rechtvaardigt – kan haar derhalve niet baten, nu het in het onderhavige geval gaat om een ander product. Dat in de marktvergunning voor irbesartan (onder dosering) is vermeld dat bij onvoldoende therapeutisch effect naast irbesartan tevens een ander anti-hypertensief middel, in het bijzonder een diureticum zoals HCTZ, kan worden toegevoegd, maakt dit niet anders. Die vermelding maakt immers niet dat de marktvergunning voor irbesartan tevens heeft te gelden als marktvergunning voor de combinatie met HCTZ en evenmin dat het combinatieproduct niet langer zou hebben te gelden als ander product in de zin van de ABC-Verordening.
ad ii) artikel 3 (c) ABC-Verordening
4.16. Onder verwijzing naar het Medeva-arrest [hier]van het HvJ EU, de conclusie van A-G Trstenjak bij dat arrest en het doel en de strekking van de ABC-Verordening stelt Teva dat het slechts mogelijk is één ABC per basisoctrooi te verkrijgen. Nu EP 511 ook als basisoctrooi is aangemerkt voor het – eerder verkregen en inmiddels vervallen – mono-ABC, is het combinatie-ABC volgens Teva nietig.
4.19. De voorzieningenrechter merkt voorts op dat algemeen wordt aangenomen dat uit het in 1995 gewezen Biogen-arrest de regel volgt dat (slechts) één ABC kan worden verkregen per product per basisoctrooi. Gelet op het feit dat het in die zaak ging om één geneesmiddel dat door meerdere basisoctrooien wordt beschermd (waarvoor verschillende octrooihouders afzonderlijke ABC’s hadden aangevraagd) en niet om een basisoctrooi dat meerdere producten in de zin van de ABC-Verordening beschermt, kan uit de door het HvJ EU gebruikte formulering dat “voor ieder basisoctrooi niet meer dan één certificaat [mag] worden afgegeven” naar voorlopig oordeel niet worden geconcludeerd dat deze regel zonder meer van toepassing is op een basisoctrooi dat meerdere producten onder bescherming stelt (zoals EP 511).
4.22. De Haagse rechtbank heeft recent het voornemen kenbaar gemaakt prejudiciële vragen voor te leggen aan het HvJ EU in twee (bestuursrechtelijke) zaken.[red. zie IEF 11581] Hoewel de feiten in beide zaken enigszins afwijken van die in het onderhavige kort geding, zou het oordeel van het HvJ EU in die zaken van belang kunnen zijn voor de thans voorliggende vraag. Gelet op het spoedeisend karakter van de onderhavige zaak en de verwachting dat een oordeel van het HvJ EU niet binnen afzienbare tijd zal worden gegeven ligt het niet in de rede dat oordeel af te wachten.
4.23. Gelet op hetgeen hiervoor is overwogen ziet de voorzieningenrechter in de onderhavige zaak geen aanleiding af te wijken van de in elk geval tot het Medeva-arrest gevolgde – en in sommige jurisdicties ook nadien gevolgde5 – bestendige lijn dat het aantal ABC’s is beperkt tot één per product per basisoctrooi. De verlening van het mono-ABC doet aan de geldigheid van het combinatie-ABC voorshands dan ook niet af.
4.26. De voorzieningenrechter stelt voorop dat het hanteren van definities, zoals de in artikel 1 van de ABC-Verordening gegeven definitie voor “product”, met zich brengt dat bij grensgevallen sprake kan zijn van situaties waarin verlening van een ABC – of juist weigering ervan6 – niet direct in overeenstemming lijkt met doel en strekking van de ABCVerordening. In de ABC-Verordening noch in de uitspraken van het Europese Hof van Justitie is evenwel een aanwijzing te vinden voor de door Teva bepleite regel dat los van toetsing aan de vereisten van artikel 3 ABC-Verordening, als aanvullend criterium heeft te gelden dat een ABC in overeenstemming dient te zijn met doel en strekking van de ABCVerordening. Nu naar voorlopig oordeel aan de vereisten van artikel 3 ABC-Verordening is voldaan, ziet de voorzieningenrechter derhalve voorshands geen aanleiding te veronderstellen dat het combinatie-ABC een nietigheidsaanval wegens strijd met de ABCVerordening in een bodemprocedure niet zou overleven.
ad iii) is EP511 nietig wegens gebrek aan inventiviteit?
4.34. De stelling van Teva dat uit de aanvrage onvoldoende van enig werkelijk technisch effect (onderbouwing van de werkzaamheid van de gevonden verbindingen) blijkt en dat onderbouwing daarvan na de datum van indiening – zoals op verzoek van de Examiner is gebeurd – ontoelaatbaar zou zijn, wordt verworpen. Naar voorlopig oordeel was het voor de gemiddelde vakman uit het octrooi (respectievelijk het prioriteitsdocument of de aanvrage) – waaronder in het bijzonder de in 2.5 hiervoor geciteerde passages – voldoende aannemelijk dat het voordelig is om (onder meer) irbesartan toe te passen bij de behandeling
van hypertensie. Het is volgens vaste (EOB-) rechtspraak niet nodig dat op de prioriteitsdatum reeds een volledige experimentele onderbouwing daarvan beschikbaar is en wordt beschreven. Dit zou anders kunnen zijn, bijvoorbeeld indien er gerede twijfel zou zijn over de vraag of het gestelde effect inderdaad met irbesartan kan worden behaald, of indien duidelijk zou zijn dat het octrooi in feite op speculatie berust. Dat irbesartan werkzaam is (gebleken), is door Teva niet gemotiveerd bestreden en van speculatie door de
octrooihouder is naar voorlopig oordeel geen sprake.
Dictum:
verbiedt Pharmachemie en Teva Nederland met onmiddellijke ingang na betekening van dit vonnis in Nederland inbreuk te maken op het combinatie-ABC (Aanvullend Beschermingscertificaat ABC 990006);
Geen dieet, geen oefeningen, toch afvallen
RCC 21 augustus 2012, dossiernr. 2012/00700 (Sweet & Slim)
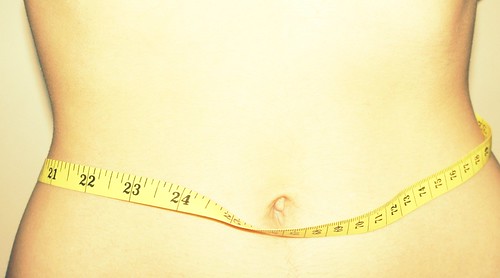 “De mooie reclamebelofte over product Sweet & Slim houdt door gebrek aan feiten geen stand.”. Preventief KAG-advies niet ingewonnen, strijd met CAG, Warenwet, Geneesmiddelenwet. Misleidende reclame, bewijslast verdeling bij de adverteerder voor een geclaimde uiting; in strijd met art. 7 Nederlandse Reclame Code (hierna: NRC).
“De mooie reclamebelofte over product Sweet & Slim houdt door gebrek aan feiten geen stand.”. Preventief KAG-advies niet ingewonnen, strijd met CAG, Warenwet, Geneesmiddelenwet. Misleidende reclame, bewijslast verdeling bij de adverteerder voor een geclaimde uiting; in strijd met art. 7 Nederlandse Reclame Code (hierna: NRC).
Voor het afslankproduct “Sweet & Slim” worden er uitingen in een huis-aan-huis folder gedaan over de speciale werking van het product om af te vallen zonder hiervoor een dieet te volgen en er oefeningen voor te doen. Het gaat hier om de volgende uiting:
“Sweet & Slim is speciaal ontwikkeld voor het bestrijden van zwaarlijvigheid, en het is zo’n krachtig werkend middel, dat ik het alleen kan voorschrijven in de allerkleinste hoeveelheden. U hoeft Geen Dieet te Volgen en U Hoeft Geen Oefeningen te Doen (…)”.
Klager heeft het product besteld. In de folder staat dat men geen dieet hoeft te volgen en uitsluitend de pillen dient te slikken om af te vallen. In de verpakking van het product zit echter een gebruiksaanwijzing waarin ook dieet-tips staan. Klager is van mening dat hij, wanneer hij een dieet volgt, geen pillen hoeft in te nemen.
De uiting is niet aan de KAG ter preventieve toetsing voorgelegd. Zou dat wel zijn gebeurd zouden er strijd met artikel 10 Code Aanprijzing Gezondheidsproducten (cfrm. 19 / 20 Warenwet) zijn. En daarbij strijd met 6 CAG en 84 Geneesmiddelenwet: - “(…)zorgt ook voor het verlagen van uw cholesterol en het vermindert ook het risico op een hartaandoening en andere organische aandoeningen”.
Voorts acht de Keuringsraad de volgende claims onaannemelijk en misleidend, en derhalve in strijd met de artikelen 7 en 35 CAG:
- “(…) gemakkelijk 15 kilo kwijtraakten in de proefperiode van 10 dagen, zonder diëten, oefeningen of een operatie”,
- “iedere proefpersoon raakte 15 kilo kwijt in 10 dagen en 0,2% van de proefpersonen viel zoveel af dat ze moesten stoppen” en
- “in dit geval is lichaamsbeweging niet noodzakelijk als hulp bij het afvallen”.
Tot slot: Adverteerder garandeert de werking van het product zoals die eveneens is verwoord in de advertentie. Het enkele innemen van de zoetstofkorrels conform de gebruiksaanwijzing leidt tot de beoogde gewichtsafname. Hoewel het volgen van een dieet derhalve niet noodzakelijk is om het beloofde resultaat te bereiken, heeft adverteerder algemene dieettips bij de verzending van zijn product ingesloten. Het is algemeen bekend dat een gezonde en evenwichtige voeding bijdraagt aan een gezonde levensstijl en er eerder toe zal bijdragen dat de beoogde resultaten behouden kunnen worden, terwijl het voedingspatroon evenwichtig blijft.
Commissie:
In de uiting wordt op verschillende plaatsen, al dan niet nadrukkelijk, meegedeeld dat het product, zonder dat men een dieet hoeft te volgen en zonder oefeningen te doen, ervoor zorgt dat men ‘flink wat overgewicht’ kwijt raakt.
Zo wordt onder meer meegedeeld: “Sweet Slim is speciaal ontwikkeld voor het bestrijden van zwaarlijvigheid, en het is zo’n krachtig werkend middel, dat ik het alleen kan voorschrijven in de allerkleinste hoeveelheden. U hoeft Geen Dieet te Volgen en U Hoeft Geen Oefeningen te Doen (…)”. Om in 10 dagen 15 kilo af te vallen, is, volgens de uiting, “alles wat u hoeft te doen”, ‘s ochtends, ’s middags en ’s avonds “één Sweet & Slim korrel” in te nemen.
Gelet op de klacht, waarin deze werking wordt betwist, lag het op de weg van adverteerder om de werking van het product aannemelijk te maken. Adverteerder heeft echter geen stukken overgelegd ter onderbouwing van de door haar geclaimde werking.
De Commissie acht de reclame-uiting in strijd met het bepaalde in artikel 7 NRC. Zij beveelt adverteerder aan om niet meer op een dergelijke wijze reclame te maken.
Nieuwe Check Voedingsclaims
 Uit de RCC-nieuwsbrief: Graag maak ik u erop attent dat er op onze Check SRC site de afgelopen maanden 4 nieuwe Checks zijn toegevoegd. De Check Telemarketing, Check Voedingsclaims, Check Reisaanbiedingen en Check Personenauto’s. Op Check! SRC kunt u lezen wat de checks precies inhouden en tevens de andere checks raadplegen. (...)
Uit de RCC-nieuwsbrief: Graag maak ik u erop attent dat er op onze Check SRC site de afgelopen maanden 4 nieuwe Checks zijn toegevoegd. De Check Telemarketing, Check Voedingsclaims, Check Reisaanbiedingen en Check Personenauto’s. Op Check! SRC kunt u lezen wat de checks precies inhouden en tevens de andere checks raadplegen. (...)
Met de Check Voedingsclaims kan worden vastgesteld welke voedingsclaims zijn toegestaan (dit is slechts een beperkt aantal) en welke alternatieve bewoordingen voor de goedgekeurde voedingsclaims mogen worden gebruikt. (...)
Wat vindt u?
SRC is bezig om de Check! SRC site meer onder de aandacht te brengen. Met deze site willen we adverteerders al in het beginstadium van het maken van reclame bijstaan met advies over hoe verantwoord reclame te maken en waar men zoal aan moet denken. We nodigen u uit een kijkje te nemen op de site Check! SRC en ons te laten weten wat u er van vindt. Daarnaast komen we graag in contact met reclamemakers die ons willen helpen onze site te optimaliseren door als ‘testgebruiker’ onze site kritisch te bekijken zodat we u in de toekomst nog beter van dienst kunnen zijn. U kunt ons dit laten weten door een email te sturen naar communicatie@reclamecode.nl.
Beroep Thuiszorg Service Nederland tegen steun HWW niet ontvankelijk
CBB 13 september 2012, LJN BX6991 (Thuiszorg Service Nederland tegen NZa)


Beleidsregel steunverlening AWBZ (CA-398). Artikel 108, derde lid, VWEU. Staatssteun. Procesbelang. Taak nationale rechter bij schending verplichting tot voorafgaande aanmelding van steunmaatregelen bij Europese Commissie.
Uit't persbericht: Het College van Beroep voor het bedrijfsleven deed op 13 september 2012 uitspraak in de zaak van Thuiszorg Service Nederland (TSN) tegen de Nederlandse Zorgautoriteit (NZa).
In maart 2009 ging Meavitagroep Den Haag failliet. Kort voor het faillissement was een stichting opgericht om de continuering van de zorg en huishoudelijke verzorging te waarborgen, namelijk Stichting Continuering Uitvoering AWBZ West (HWW).
NZa verleende aan HWW ruim € 22 miljoen steun. TSN keerde zich tegen die steunverlening met als argument dat het om staatssteun zou gaan. De beroepsprocedure is op verzoek van TSN in afwachting van de verkoop van HWW een aantal keren uitgesteld. TSN zwakte haar eis in de loop van de procedure af en vroeg uiteindelijk NZa de keuze te laten tussen aanmelding van de steun aan de Europese Commissie en terugvordering. Als TSN gelijk zou krijgen, dan kan dat verzoek niet worden ingewilligd zonder in strijd te komen met de taak van de nationale rechter om de toestand vóór de steunverlening te herstellen. Het doel dat TSN met het beroep voor ogen staat, kan niet worden bereikt. Het College verklaart het beroep van TSN daarom niet-ontvankelijk.
5.8 Het vorenstaande leidt het College tot de slotsom dat het verzoek van Thuiszorgservice, zoals gewijzigd in haar brief van 22 december 2011, door het College - indien zou worden geoordeeld dat verweerster niet aangemelde staatssteun heeft verleend aan HWW - niet kan worden ingewilligd zonder in strijd te komen met de uit de hiervoor onder 5.4 omschreven taak van de nationale rechter voortvloeiende verplichting om zodanige maatregelen te nemen dat de toestand vóór de steunverlening wordt hersteld. In zoverre wordt in dit geval derhalve niet voldaan aan het vereiste, dat het doel dat Thuiszorgservice met het instellen van het beroep voor ogen staat, met het beroep moet kunnen worden bereikt. Dit stelt de vraag aan de orde of Thuiszorgservice voor het overige nog een voor de ontvankelijkheid toereikend procesbelang heeft bij het voeren van deze procedure.
Een rechtens relevant belang bij het voeren van deze procedure is, zoals volgt uit hetgeen hiervoor is overwogen, naar zijn aard aanwezig, indien het beroep geacht kan worden ertoe te strekken dat de rechter de nodige maatregelen neemt die ertoe leiden dat een, wegens niet aanmelding, onwettige steunmaatregel ongedaan wordt gemaakt, namelijk doordat verzocht wordt de uitvoering van een voorgenomen steunmaatregel te schorsen of doordat de terugvordering van verleende steun wordt geëist. Het vereiste belang ontbreekt echter naar het oordeel van het College indien het voeren van de procedure geacht moet worden uitsluitend gericht te zijn op het verkrijgen van een oordeel van de nationale rechter over de - in dat geval als academisch te kwalificeren - vraag of een besluit wel of niet staatssteun betreft, waarbij het vervolgens aan de discretie van het betrokken bestuursorgaan wordt gelaten welke consequenties het daaruit trekt, of gericht te zijn op het behouden van een versterkte onderhandelingspositie bij, in dit geval, het bieden op de hiervoor bedoelde marktplaats. Gezien de inhoud van haar verzoek, zoals door haar nader toegelicht, is het College van oordeel dat het slechts laatstbedoelde belangen zijn waarop de onderhavige procedure van Thuiszorgservice nog is gericht. Gesteld noch gebleken is dat Thuiszorgservice nog om andere redenen een voldoende belang heeft bij een inhoudelijk oordeel over het bestreden besluit. Het beroep dient derhalve wegens het ontbreken van procesbelang niet-ontvankelijk te worden verklaard.
Samenwerking wetenschappelijk advies tussen CBG en CVZ
Uit't persbericht: Vanaf 1 september 2012 is het mogelijk om gelijktijdig, dus binnen een enkele procedure, een wetenschappelijk advies aan te vragen bij zowel het College ter Beoordeling van Geneesmiddelen (CBG, ten behoeve van registratie) en als het College voor Zorgverzekeringen (CVZ, ten behoeve van vergoeding). Dit kan voor firma’s relevant zijn, bijvoorbeeld bij de opzet van een klinische studie (fase 3) zodat deze kan voldoen aan de richtlijnen voor registratie en vergoeding. Ieder neemt uitdrukkelijk verantwoording voor dat deel van het advies dat binnen de competentie van respectievelijk het CBG of het CVZ valt . De samenwerking zal na een jaar worden geëvalueerd.
Als u van deze procedure gebruik wilt maken, dan kunt u het reguliere aanvraagformulier van de CBG-site voor wetenschappelijk advies gebruiken en de specifieke vraag (inclusief standpunt van de aanvrager) voor CVZ daar aan toe voegen. Geeft u dit duidelijk aan op het aanvraagformulier en/of de begeleidende aanbiedingsbrief (mail). Alle verdere correspondentie loopt ook via het CBG (case@cbg-meb.nl o.v.v. zaaknummer).
Meer informatie
Hoe dien ik een wetenschappelijk advies in?
Against All Dots
WIPO Panel Decision (Christopher J. Pibus) 9 augustus 2012, D2012-1117 (Dotmed.com tegen Hexap c.s.)
![]() Met een analyse door Thijs van den Heuvel, Bird & Bird LLP.
Met een analyse door Thijs van den Heuvel, Bird & Bird LLP.
Als randvermelding, eerder gepubliceerd als IE-Forum nr. IEF 11744. Domeinnaamrecht. Ter promotie van de kandidatuur voor de gTLD zijn de registraties van de verwarringwekkend overeenstemmende dotmed-aanvragen toegestaan.
Ruim een maand geleden schreef ik over de aanstaande introductie van de nieuwe generieke topleveldomeinnamen (“gTLDs”) IEF 11623 [titel: Meer .love dan .sex....]. In een recente WIPO-procedure staat DOTMED.COM INC. (hierna: Dotmed) tegenover Hexap en Promopixel SARL (hierna Hexap), één van de aanvragers van een nieuwe gTLD. Dotmed handelt in nieuwe en gebruikte medische apparatuur en onderhoudt hiervoor een veiling website (zie dotmed.com). Dotmed is onder meer houdster van de merken DOTMED en DOTMED.COM.
Hexap is verbonden aan het webhosting bedrijf Promopixel en heeft recentelijk bij het ICANN een aanvraag gedaan voor het generieke topleveldomein (gTLD) < .med >. Zij heeft voor deze gTLD concurrentie van drie andere aanvragers. Hexap heeft naast deze aanvraag de volgende domeinnamen geregistreerd: < aboutdotmed.com >, < thedotmed.com > en < supportdotmed.com > .
Dotmed klaagt dat deze domeinnamen verwarringwekkend overeenstemmend zijn met haar merk DOTMED. De geschillenbeslechter van het WIPO is het op dit punt met Dotmed eens. De enkele toevoeging van “about”, “the” en “support” aan het merk DOTMED maakt geen relevant verschil. Hexap stelt echter dat zij een legitiem belang heeft bij de geregistreerde domeinnamen. Zij heeft de domeinnamen namelijk geregistreerd om haar aanvraag voor de gTLD <.med> te promoten. Op < aboutdotmed.com > wordt gedetailleerd uitleg gegeven over het “.med Project” en de kandidatuur van Hexap voor deze gTLD.
Hexap stelt verder dat gezien het feit dat op dit moment geen enkele domeinnaam een <.> (dot) kan omvatten als deel van de naam, zij wel gedwongen was om de fonetische variant te gebruiken, te weten “dotmed”. Het WIPO gaat hier in mee en overweegt dat het voor Hexap niet mogelijk was om de domeinnamen <about.med.com>, <support.med.com> en <the.med.com> te registreren, en dat het daarom redelijk is dat zij de punt <.> voluit schreef als <dot>.
Dit is enigszins opmerkelijk, omdat het wel degelijk mogelijk is om de domeinnamen < about.med.com > etc. te gebruiken. Dat Hexap hiertoe niet in staat was komt niet doordat domeinnamen geen <.> kunnen omvatten - zoals Hexap had gesteld en door het WIPO werd geaccepteerd - maar doordat het (second level) domein <med.com> al in handen is van een derde partij (Trentville Corporation N.V).
De geschillenbeslechter van het WIPO komt uiteindelijk tot de conclusie dat Hexap de domeinnamen inderdaad heeft geregistreerd om het publiek te kunnen informeren over haar <.med> gTLD project en dat zij daarbij een legitiem belang heeft. Hierbij speelt het feit dat Hexap drie concurrenten heeft voor de betreffende gTLD ook een rol. De klacht van Dotmed wordt afgewezen, en zij moet - in ieder geval voorlopig – met lede ogen toezien dat Hexap de drie domeinnamen blijft gebruiken.
Onvoldoende om onmiskenbaar onrechtmatig aan te merken
Vzr. Rechtbank 's-Gravenhage 12 september 2012, LJN BX7092 (Tandartsen c.s. tegen de Staat)

Aanwijzing inhoudende dat het 'Experiment vrije prijsvorming mondzorg' [hier / beleidsregel BR/CU-7058] wordt stopgezet en dat met ingang van 1 januari 2013 maximumtarieven worden ingevoerd. Eisers vorderen dat de Aanwijzing buiten werking wordt gesteld en/of de Staat te verbieden de Aanwijzing te effectueren. Aan eisers moet worden toegegeven dat de besluitvorming tot stopzetting van het Experiment vragen oproept over de consistentie van het gevoerde beleid. Uit het aanvankelijk door de Minister ingenomen standpunt, zoals verwoord in de onder 1.11 vermelde brief en het onder 1.12 vermelde overleg met de Tweede Kamer, kan worden afgeleid dat ten tijde van de totstandbrenging van de Aanwijzing niet was voldaan aan de vooraf voor tussentijdse beëindiging van het Experiment geformuleerde voorwaarden. Op zichzelf is het ook juist dat de Minister staatsrechtelijk niet verplicht was de motie-Kuiken c.s. uit te voeren. Dit een en ander is evenwel onvoldoende om te komen tot het oordeel dat de Aanwijzing als onmiskenbaar onrechtmatig moet worden aangemerkt.
In zijn algemeenheid geldt immers dat de Staat - in dit geval de Minister - een ruime mate van vrijheid heeft om het gevoerde beleid te wijzigen, ook als daardoor eerdere verwachtingen of vooruitzichten teniet worden gedaan. Dit is slechts anders indien het oordeel zich opdringt dat de Minister - in aanmerking genomen de bij het besluit betrokken belangen - in redelijkheid niet tot het betreffende besluit heeft kunnen komen. Naar het oordeel van de voorzieningenrechter is daarvan geen sprake, redengevend daarvoor is het volgende. Met de invoering van het Experiment is beoogd te bezien of vrije prijsvorming in de mondzorg zou leiden tot een betere prijs-kwaliteitverhouding voor de consument. Mede gelet op de weerstand tegen deze vrije prijsvorming is bij de invoering van het experiment als voorwaarde gesteld dat het experiment tussentijds beëindigd kon worden in geval van - onder meer - ontoelaatbare prijsstijgingen. In de Marktscan is vastgesteld dat de tarieven in 2012 ten opzichte van 2011 zijn gestegen, hetgeen overigens wordt bevestigd door de in deze procedure door eisers overgelegde halfjaarcijfers. Ook indien in aanmerking wordt genomen dat de omvang van de prijsstijging en het ontoelaatbare karakter ervan voor discussie vatbaar zijn - een discussie die het bestek van dit kort geding ver te buiten gaat - kan in het kader van de Wmg in die prijsstijging een gerechtvaardigde aanleiding worden gevonden het Experiment, conform de uitdrukkelijke wens van de Tweede Kamer, tussentijds te beëindigen. Hoewel evident is dat de belangen van eisers door die tussentijdse beëindiging worden getroffen, is niet gesteld of gebleken dat de door eisers gestelde belangen daardoor onevenredig worden getroffen. Niet valt uit te sluiten dat de ten behoeve van het Experiment gepleegde investeringen ook na 1 januari 2013 hun waarde zullen behouden.
Fruity Dutch advertisements: how misleading are they?
![]() Als randvermelding. In het september nummer van European Food and Feed Law tijdschrift is het "Country Report" voor Nederland verschenen van de hand van Ebba Hoogenraad en Christine Fontaine, Hoogenraad & Haak advertising + IP advocaten.
Als randvermelding. In het september nummer van European Food and Feed Law tijdschrift is het "Country Report" voor Nederland verschenen van de hand van Ebba Hoogenraad en Christine Fontaine, Hoogenraad & Haak advertising + IP advocaten.
Lees hier de bijdrage "Fruity Dutch advertisements: how misleading are they?"














