Prejudiciële vragen betreffende het begrip geneesmiddel
Prejudiciële vragen gesteld aan HvJ EU 15 mei 2013, zaak C-267/13 (Nutricia) - dossier![]() Douanerecht. Sondevoeding aanmerking als geneesmiddel. Prejudiciële vragen betreffende Verordening (EG) 1549/2006 van de Commissie tot wijziging van Verordening (EEG) nr. 2658/87 van de Raad met betrekking tot de tarief- en statistieknomenclatuur en het gemeenschappelijk douanetarief.
Douanerecht. Sondevoeding aanmerking als geneesmiddel. Prejudiciële vragen betreffende Verordening (EG) 1549/2006 van de Commissie tot wijziging van Verordening (EEG) nr. 2658/87 van de Raad met betrekking tot de tarief- en statistieknomenclatuur en het gemeenschappelijk douanetarief.
Verzoekster heeft op aanvraag vijf bindende tariefinlichtingen (op grond van artikel 12 CDw) van de Inspecteur gekregen waartegen bezwaar is gemaakt dat (in één verweerschrift) is afgewezen. De Rechtbank Haarlem verklaart het door verzoekster ingestelde beroep ongegrond, het hof Amsterdam bevestigt het arrest waarna verzoekster in cassatie gaat.
De zaak gaat over vijf soorten sondevoeding. Verzoekster heeft gevraagd deze producten in te delen onder post 3004 50 10 van de GN, maar de Inspecteur deelt in onder 2202 90 10 (‘andere alcoholvrije dranken’). De omschrijving van de producten staat bovenaan pagina 3 van de verwijzingsbeschikking. De indeling heeft plaatsgevonden na onderzoek door het Douanelab en de onderzoeksresultaten zijn op het sacharosegehalte na gelijk, zie pagina 4. Het hof Amsterdam oordeelt dat de producten moeten worden ingedeeld onder 2202 90 10. Niet onder post 3004 omdat indeling als ‘geneesmiddel voor therapeutisch of profylactisch gebruik’ slechts mogelijk is indien een product beschikt over nauwkeurig omschreven therapeutische en profylactische kenmerken waarvan de werking zich richt op welbepaalde functies van het menselijk lichaam. Het door verzoekster geclaimde voorkomen en bestrijden van ondervoeding voldoet daar volgens het hof Amsterdam niet aan
Verzoekster stelt in haar middelen dat het hof Amsterdam teveel de nadruk legt op eventuele therapeutische of profylactische kenmerken van de producten en ten onrechte het therapeutische of profylactische gebruik ervan buiten beschouwing laat. Zij meent dat er voldoende grond is voor indeling in post 3004 GN (geneesmiddelen). Voor wat betreft de voorgestelde indeling onder post 2202 van de GN stelt zij dat hiervoor vereist is dat het product bestemd is om te worden gedronken en dat is voor haar producten niet het geval.
De verwijzende Nederlandse rechter (HR) vindt in jurisprudentie van het HvJEU dat van doorslaggevend belang voor indeling onder 3004 ‘geneesmiddelen’ is dat het product therapeutische of profylactische eigenschappen of kenmerken heeft, waarvan de werking zich richt op welbepaalde functies van het menselijk organisme, of die kunnen worden gebruikt ter voorkoming of behandeling van een ziekte of een aandoening. In het geval van verzoekster zou dit indeling onder 3004 in de weg staan omdat de werking van de producten zich niet richt op behandeling of voorkoming van bepaalde ziekten of aandoeningen. Verder vraagt de rechter zich af wat onder ‘dranken’ in de GN-code wordt verstaan. Hij stelt de volgende vragen aan het HvJEU:
1. Moet het begrip 'geneesmiddel' in de zin van post 3004 van de GN zo worden uitgelegd dat daaronder mede zijn begrepen voedingspreparaten als de onderhavige producten, die uitsluitend bestemd zijn om onder medisch toezicht enteraal (door middel van een maagsonde) te worden toegediend aan personen die wegens ziekte of aandoening medisch worden behandeld en in het kader van de bestrijding van die ziekte of aandoening de producten krijgen toegediend ter bestrijding of voorkoming van ondervoeding?
2. Moet het begrip 'dranken' in de zin van post 2202 van de GN zo worden uitgelegd dat daaronder zijn begrepen vloeibare voedingsmiddelen als de onderhavige producten, die niet zijn bestemd om te worden gedronken maar om enteraal (door middel van een maagsonde) te worden toegediend?
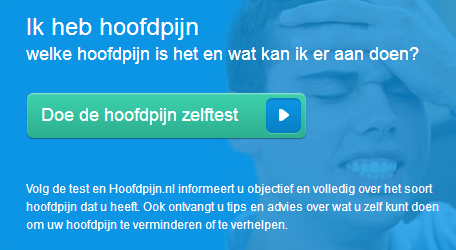 Medicijnen. Vorm van beïnvloeding. Kennelijke doel. Aanbeveling. Het betreft een radiospotje met tag-on (hierna ook: radiocommercial), waarin het volgende wordt gezegd: “Wordt jouw dag regelmatig verpest door hoofdpijn? En helpen de pijnstillers van de drogist niet? Misschien heb je migraine. Doe de hoofdpijntest op hoofdpijn.nl. Je huisarts kan specifieke medicijnen tegen migraine voorschrijven die jouw dag weer mooi kunnen maken”. Tag-on: “Laat je dag niet verpesten door migraine. Meer weten? Ga naar hoofdpijn.nl”.
Medicijnen. Vorm van beïnvloeding. Kennelijke doel. Aanbeveling. Het betreft een radiospotje met tag-on (hierna ook: radiocommercial), waarin het volgende wordt gezegd: “Wordt jouw dag regelmatig verpest door hoofdpijn? En helpen de pijnstillers van de drogist niet? Misschien heb je migraine. Doe de hoofdpijntest op hoofdpijn.nl. Je huisarts kan specifieke medicijnen tegen migraine voorschrijven die jouw dag weer mooi kunnen maken”. Tag-on: “Laat je dag niet verpesten door migraine. Meer weten? Ga naar hoofdpijn.nl”. This guidance document addresses a number of questions which marketing authorisation holders (MAHs) may have on post-authorisation procedures. It provides an overview of the Agency’s position on issues, which are typically addressed in discussions or meetings with MAHs in the post-authorisation phase.
This guidance document addresses a number of questions which marketing authorisation holders (MAHs) may have on post-authorisation procedures. It provides an overview of the Agency’s position on issues, which are typically addressed in discussions or meetings with MAHs in the post-authorisation phase.%20wikimedia.png) Omdat de prijsvorming van geneesmiddelen ook beïnvloed kan worden door de hoogte van de vergoedingslimieten heb ik tijdens het algemeen overleg van 12 december 2012 aangegeven in het standpunt op het rapport ook in te gaan op de functie en het effect van het Geneesmiddelenvergoedingssysteem (Gvs) op de prijsvorming. Inmiddels is ook een aantal VSO-vragen gesteld over de Wgp, het Gvs, en het Noorse systeem van prijsbeheersing in relatie tot het rapport van Conquaestor. Zoals al eerder schriftelijk toegezegd2, zal ik deze vragen eveneens beantwoorden in deze brief. Voordat ik hiertoe over ga zal ik aangeven hoe de prijsvorming in Nederland en Noorwegen plaatsvindt en wat de verschillen zijn tussen beide stelsels van prijsvorming. In de bedoelde VSO’s zijn ook vragen gesteld over onderwerpen die geen directe relatie hebben met de hierboven genoemde onderwerpen. Die vragen beantwoord ik niet in deze brief. Ik doe dat op een later moment alsnog, als alle informatie die voor de beantwoording van de resterende vragen nodig is, is ingewonnen.
Omdat de prijsvorming van geneesmiddelen ook beïnvloed kan worden door de hoogte van de vergoedingslimieten heb ik tijdens het algemeen overleg van 12 december 2012 aangegeven in het standpunt op het rapport ook in te gaan op de functie en het effect van het Geneesmiddelenvergoedingssysteem (Gvs) op de prijsvorming. Inmiddels is ook een aantal VSO-vragen gesteld over de Wgp, het Gvs, en het Noorse systeem van prijsbeheersing in relatie tot het rapport van Conquaestor. Zoals al eerder schriftelijk toegezegd2, zal ik deze vragen eveneens beantwoorden in deze brief. Voordat ik hiertoe over ga zal ik aangeven hoe de prijsvorming in Nederland en Noorwegen plaatsvindt en wat de verschillen zijn tussen beide stelsels van prijsvorming. In de bedoelde VSO’s zijn ook vragen gesteld over onderwerpen die geen directe relatie hebben met de hierboven genoemde onderwerpen. Die vragen beantwoord ik niet in deze brief. Ik doe dat op een later moment alsnog, als alle informatie die voor de beantwoording van de resterende vragen nodig is, is ingewonnen. Mevrouw Dijkstra (D66) heeft tijdens het VAO een motie (
Mevrouw Dijkstra (D66) heeft tijdens het VAO een motie ( Contractenrecht. Zorgverzekeraar. Verbod kosten verstrekking en/of toediening aan verzekerden.
Contractenrecht. Zorgverzekeraar. Verbod kosten verstrekking en/of toediening aan verzekerden.













